Homogeneous Nucleation
The aim of the theme was to seek improved understanding of the broadly interpreted phenomenon of “homogeneous nucleation”. This encompasses any nucleation which is not initiated at the interface between a parent phase and a surface/nucleant. Within that definition we recognise that bulk solutions are often far from homogeneous, and that surfaces can induce inhomogeneities in solution as well as acting as nucleation sites themselves.
We have built minimal physics models which capture non-trivial nucleation and growth mechanisms to improve our understanding of multi-component systems and competing nucleation products. We have explored the kinetic and thermodynamic assumptions of classical nucleation theory and showed how these can limit the accuracy of quantities computed from computer simulations at the molecular scale. Improved and accelerated simulation methods have been explored, based on enhanced quantitative descriptions of nucleation pathways. Finally, we have pioneered novel ways of measuring induction times to nucleation of amorphous calcium carbonate via electrochemical methods.
Minimal models for non-trivial nucleation behaviour
Two class of simple model have been developed. The first is based on the Ising lattice gas – the most simple model of solute precipitation in two dimensions. We add defects to this model, representing impurities in solution or on a nucleating substrate. These can be static or dynamic, with markedly different behaviour depending on the effective mobility of the impurities [reference a]. We have further generalised the model to explore the types of nucleation and growth mechanism exhibited when impurities are attracted to either solute or solution particles [reference c] leading to the behaviour map below.
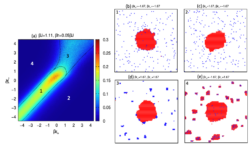
Figure 1: Nucleation behaviour map for impurities which act as (1) surfactant, lowering the interfacial free energy of nuclei (2) solution stabilisers, (3) spectator clusters or (4) nucleants depending on the energy of interaction with solvent and solute particles. See reference [c] for details.
In a second class of model, we consider two types of solute species which form different space-filling structures. Interactions between solute and solvent are carefully tuned such that nucleation of the two structures is in close competition and can hence lead to mixtures, or complex nucleation pathways in which the less stable structure nucleates first and is eventually out-competed by the slightly more stable structure [reference b].

Figure 2: Sequence of snapshots during nucleation and growth in a system for which the red dimer phase is most stable, but in which the metastable blue phase nucleates more rapidly. Time progresses from left to right. Simulations are performed at very high dimer chemical potential and hence multiple nuclei of each phase are observed.
This model is finding utility for understanding situations where the nucleated phase sensitively depends on external conditions, such as under confinement or where the mobility of one type of solute is restricted.
References:
[a] Mandal D. and Quigley D. “Nucleation rate in the two-dimensional Ising model in the presence of random impurities”, Soft Matter (2021) 17(38), 8642-8650
[b] Mandal D. and Quigley D. “Kinetic control of competing nuclei in a dimer lattice-gas model”, J. Chem. Phys. (2022) 157, 214501.
[c] Mandal D. and Quigley D. “Mapping the influence of impurities with varying interaction strength on nucleation in the 2D Ising model” [preprint].
Improved calculations of nucleation behaviour from atomistic simulations
Minimal physics models such as the above are inexpensive to simulate but lack the chemical specificity to make quantitative predictions about real systems. Detailed atomistic models are however much more expensive, requiring advanced accelerated simulations techniques and some loss of fidelity as discussed in a recent review from our team [reference f].
Even the prototypical example of sodium chloride precipitation can exhibit non-trivial behaviour when probed at the appropriate level of detail. We have shown that nucleation can proceed via either the classical single-step where ordered nuclei form directly, or via a two-step mechanism where disordered aggregates are the first-formed species which subsequently crystalise. Advanced Markov state models were used to predict the nucleation rate for these pathways [reference d].
![Figure 3: [A] Schematic demonstrating possible one and two-step nucleation pathways revealed from detailed atomistic simulations of sodium chloride precipitation [d]. An example pre-critical cluster from along the two-step pathways is shown in [B] coloured by solute-solute local order parameter from low (red) to high (blue).](https://realworldcrystals.leeds.ac.uk/wp-content/uploads/sites/41/2024/02/TA1-fig-3-250x123.gif)
Figure 3: [A] Schematic demonstrating possible one and two-step nucleation pathways revealed from detailed atomistic simulations of sodium chloride precipitation [d]. An example pre-critical cluster from along the two-step pathways is shown in [B] coloured by solute-solute local order parameter from low (red) to high (blue).
Simulations such as these are critically dependent on our ability to quantify progress along a nucleation and growth pathway from the current molecular positions (coordinates) as a simulation evolves. Such pathways are necessarily described via collective coordinates (CVs) – a representation in fewer dimensions than the many molecular degrees of freedom in the system. Poor choice of CV can lead to inaccurate predictions and misinterpretation of simulations. We have developed a variational approach to assessing the suitability of CV choices and tested this in the context of colloidal crystal nucleation [reference e]. This will enhance our ability to perform future accelerated simulations with improved accuracy and reliability.
References:
[d] Finney A.R. and Salvalaglio M. “Multiple pathways in NaCl homogeneous crystal nucleation”, Faraday Discussions (2022) 235, 56-80
[e] Finney A.R. and Salvalaglio M. “A variational approach to assess reaction coordinates for two-step crystallization”, J. Chem. Phys. (2023) 158, 094503
[f] Finney A.R. and Salvalaglio M. “Molecular simulation approaches to study crystal nucleation from solutions: Theoretical considerations and computational challenges”, WIREs Comput Mol Sci (2024) 14(1) e.1697
Challenging the assumptions of classical nucleation theory
Alternative approaches to extracting quantitative information on nucleation processes from molecular simulations are reliant on extracting parameters from the simulation that can be related to parameters in classical nucleation theory (CNT) a semi-quantitative theory from which (for example) one can calculate the rate at which stable crystals are formed per unit volume and unit time. The limits of this and other approaches have been reviewed by our team [reference g].
We have focussed on two questions key to accuracy via this approach. Firstly, are simulations which are “seeded” with pre-formed nuclei to extract CNT parameters representative of realistic progression from a supercooled fluid or supersaturated solution to a stable crystal? A PhD project aligned to the programme grant [reference h] has explored the key CNT assumption that an ensemble of shrinking and growing ideal crystal seeds are in quasi-equilibrium with the surrounding fluid. However, a detailed set of unbiased simulations have revealed that “real world” crystal nuclei have little in common with ideal crystalline seeds in terms of shape, energy and propensity to reach macroscopic size. They also grow/shrink at a rate faster than relaxation to quasi-equilibrium at each cluster size [reference j] suggesting that a free energy description of the process is inappropriate.
Secondly, can the dynamics of the collective variable (CV – see above) be usefully assumed to be Markovian. This property of a process implies that the next “step” along a nucleation pathway depends only on the current configuration of the system and not its history, i.e. the process has no “memory”. Using traditional CVs common in the literature, we have demonstrated [reference h] that non-Markovian descriptions of how these evolve have a higher statistical likelihood of capturing the dynamics of seeded nucleation trajectories. Related work [reference i] has demonstrated using a minimal model that apparent non-Markovian behaviour can be the result of reducing a complex process to a single CV rather than being intrinsic to the process that CV is attempting to describe.
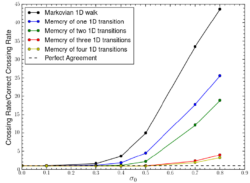
Figure 4: Illustration of non-Markovian random walks in one dimension with increasing depth of memory of previous transitions to reproduce the underlying Markovian kinetics in two dimensions. Adding memory improves the one-dimensional description of the underlying two dimensional but memory-less process. This is analogous to a non-Markovian description of a nucleation pathway described in terms of CVs where the underlying molecular dynamics is memory-less.
Both of these questions have fed into new ongoing PhD projects which will develop improved descriptions of nucleation pathways and exploit the use of “ideal” reaction coordinates for which Markovian descriptions can be shown to exist.
References:
[g] Blow K.E., Quigley D. and Sosso G.C. “The seven deadly sins: When computing crystal nucleation rates, the devil is in the details”, J. Chem. Phys. (2021) 155(4), 040901.
[h] Devonport, Craig (2020) “Thermalisation and memory in crystal nucleation”. Doctoral thesis, University of Warwick.
[i] Broad, Alexander (2023) “Tuning the Growth and Mechanical Properties of Calcite Using Impurities: Insight from Molecular Simulation”. Doctoral thesis, University College London.
[j] Blow K.E, Sosso G.C. and Quigley D. “Statistical analysis of seeded vs unbiased trajectories in crystallization from the Lennard-Jones melt” [in preparation].
Measuring nucleation times in nanopipettes
Simulations are only as good as the experimental data they have been validated against. The project has contributed to the field of experimental measurement of nucleation kinetics via development of a novel electrochemical mixing technique within a nanopipette. Formation of precipitate from solution blocks the transport of electrical current across the pipette and hence signals when a nucleus of known size has been formed. When combined with finite element modelling the nucleation induction time can be predicted as a function of supersaturation.
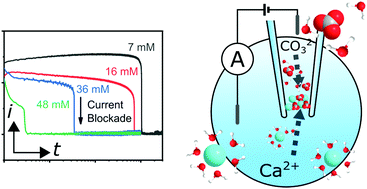
Figure 5: Left: Current against time showing the formation of a blockage and the corresponding drop in current for four solution concentrations from which amorphous calcium carbonate (ACC) is precipitated. Right: cartoon schematic of the experimental setup in which a potential different leads to an ion current between anode and cathode until the nanopipette is blocked.
When applied to precipitation of amorphous calcium carbonate [reference k] analysis indicates that neither dehydration of CO3 nor deprotonation of HCO3- is the rate limiting step to formation, information which could not be inferred via previous techniques.
References:
[k] Morris P.D., McPherson I.J., Meloni G.N. and Unwin P.R. “Nanoscale kinetics of amorphous calcium carbonate precipitation in H2O and D2O” Phys. Chem. Chem. Phys. (2020) 22, 22107-22115.